
Newsletter Articles
Gait abnormalities overlapping with ataxia in patients with LSDs
Relationship between Immune Biomarkers and Bone Pathology in type 1 Gaucher disease
Real-world effectiveness and safety of velaglucerase alfa in pediatric patients with Gaucher disease
GRIDs Symposium 2021 Highlights
Inflammatory pathways and cardiac growth factors associated with Fabry disease cardiomyopathy
A plasma biomarker for osteopenia and osteoporosis in Gaucher disease
Ambroxol decreases heparan sulfate levels and improves lysosomal functions in vitro in MPS III patient-derived primary cell lines
Cellular and biochemical response to chaperone versus substrate reduction therapies in neuropathic Gaucher disease
Lysosomal Storage Disease Quarterly CME/CE Webinar Series

Letter from the Director
Ozlem Goker-Alpan, MD
Unfortunately, before we graduated from the program, the Gene therapy fellowship was closed, and there were no Gene therapy trainees. The series of unfortunate events known to most in the field halted this promising therapy for 15 years or more, but today is the dawn of Gene therapy (GT).
GT is already approved by the FDA for two disorders, and the number of current active trials at different phases are 68 in the US and 28 in Europe in 2021.
When I started my fellowship, clinical geneticists were mainly diagnosticians. We used to describe and measure every single feature. Counting the whorls and loops of the fingerprints was my favorite. We knew too much but could do too little for our patients.
The development of Enzyme Replacement Therapy (ERT) led to the concept of Interventional Genetics. Cerezyme was approved by the FDA in May 1994, and we infused our first adult patient with Gaucher disease (GD) with the commercial ERT product with the pride of the greatest achievement.
For Lysosomal disorders, the 2000s were mainly the era of ERT or biologics. In the 2010s, the small molecule trials led to the development of effective Substrate reduction and Chaperone therapies. 2020 and beyond is the era of gene therapy. While we know current GT protocols may not offer an ultimate cure for a genetic disorder, it is the modality that comes closest to one.
We may need to wait another decade or more for CRISPR. In that time, there will be more information gathered and more GT products on the market. When I see the babies with GD2 are treated successfully, I will then be able to retire.

Gait abnormalities overlapping with ataxia in patients with LSDs
By Ozlem Goker-Alpan, MD
ATAXIC GAIT ALSO IS OBSERVED IN NEUROPATHIC GAUCHER DISEASE. THE MAJORITY OF PATIENTS MAY PRESENT WITH MORE THAN ONE MOVEMENT DISORDER.
Gait assessment is an important part of evaluation of a patient with a suspected Lysosomal disorder. Gait abnormalities and movement disorders are a significant clinical problem in lysosomal storage diseases (LSD) and account for a significant morbidity. The most common LSDs associated with a movement disorder are Niemann-Pick disease type C (NPC), neuronal ceroid lipofuscinosis (NCL), and mucopolysaccharidoses. The most common movement disorder is ataxia, followed by rest tremor, dystonia, and myoclonus. Ataxic gait is also observed in neuropathic Gaucher disease. The majority of patients may present with more than one movement disorder. In NPC and NCL, brain MRI changes include diffuse cerebral volume loss, white matter abnormalities with thinning of the corpus callosum, and cerebellar atrophy. While in GD, the MRI findings could be nonspecific. Here we present two patients, one adult with NPC and a toddler with GD3 with lower limb spasticity (spastic paraplegia) in addition to their ataxic gait.
A 3-year-old male with a history of Gaucher disease, splenomegaly, and congenital hearing loss was referred for "abnormal gait." GBA genotype was R463(502)C/ RecNciI. He had a wide-based gait, toe walking, which was more pronounced on the right. There was spasticity of the lower limbs, with the right leg circumventing on examination. An NGS Spastic paraplegia panel demonstrated a VOUS in KIF1A.
A 33-year-old male patient was referred for further evaluation. He had ataxia, balance issues, swallowing difficulties, and abnormal speech since teenage years, but for the past few years, he had progressive "tightness" in his lower legs. On examination, he had a mild vertical saccade abnormality, dysarthric speech, wide-based gait, and spasticity of the lower legs. An NGS Ataxia panel revealed one pathogenic variant in NPC1 and a VOUS in KIF1A.
Lower extremity weakness and spasticity are the predominant symptoms of inherited or hereditary spastic paraplegia (HSP). While the presentation may be at any age, the gait impairment that begins after childhood usually worsens very slowly over many years. Gait impairment that begins in infancy and early childhood may not worsen significantly as well. There are more than 50 genetic types of HSP. HSP affects individuals of diverse ethnic groups, with a prevalence estimated ranging from 1.2 to 9.6 per 100,000 [39, 70, 77, 154, 185]. Variants in family 1 kinesin (KIF1A), which encodes a kinesin axonal motor protein, have been described to cause variable neurological manifestations. Recessive missense variants have led to spastic paraplegia, and recessive truncation mutations are associated with sensory and autonomic neuropathy. De novo missense variants cause developmental delay or intellectual disability, cerebellar atrophy, and variable spasticity. More recently, variants in KIF1A have also been described in a few cases with autosomal dominant spastic paraplegia. The identification of KIF1A loss-of-function variants suggests haploinsufficiency as a possible mechanism in autosomal dominant spastic paraplegia (SPG30).
More broadly, on the more severe end of the spectrum, KIF1A gain of function mutations are associated with a spectrum of a progressive neurological disorder. KIF1A-related disorders (KRD or KAND) are first described in 2011, and the phenotypic spectrum has subsequently expanded to encompass a range of central and peripheral nervous system involvement. KIF1A Associated Neurological Disorder (KAND) is a severe and rare neurodegenerative disorder with a progressive course. KAND is caused by gain of function mutations in KIF1A. The condition can affect both the brain and body, such as the eyes, muscles, and nerves. There is a wide range of symptoms that appear at birth or in early childhood. Mutations in KIF1A can cause: cognitive impairment; cerebellar atrophy; ataxia; spastic paraplegia; optic nerve atrophy; cortical vision impairment; peripheral neuropathy, and epilepsy.
It is important to continue the detailed assessment of gait in patients with LSDs, as it may give clues not only for co-morbidities such as Parkinson disease in GD but also to identify other co-existing genetic conditions that may warrant additional work-up.
Ebrahimi-Fakhari, et al. Mov Disord Clin Pract, 2018; 149-155
Nemani et al, J Perigh Ner Sys 2020,; 117-240
"Kif1a Associated Neurological Disorder." KIF1A, 12 Dec. 2021, https://www.kif1a.org/kif1a-gene/kif1a-associated-neurological-disorder/.

Relationship between Immune Biomarkers and Bone Pathology in type 1 Gaucher disease
By Lauren Noll
Gaucher disease (GD) is the most common lysosomal storage disorder. GD is caused by mutations in the GBA gene that result in a deficiency of the lysosomal enzyme Glucocerebrosidase (GCase). There are three types of Gaucher disease, two of which (GD 2 and 3) are characterized by neuronopathic manifestations. GD type 1 (GD1) is the most common and is characterized by the absence of central nervous manifestations. Bone-related issues account for the largest unmet medical need in GD1 patients.
Over the course of 2 years, patients were observed every six months. They had blood taken and provided a urine sample at each visit, and at one visit over the course of the two years, a pain questionnaire was completed. The samples were analyzed by looking at specific biomarkers in the blood in hopes to determine if these specific biomarkers were directly related to bone pain and/or disease.
Preliminary results have shown some of the following. Osteoclasts are indicators of bone resorption. Bone resorption is the destruction of bone tissue that promotes bone loss. TRAP5B is a marker showing the active number of osteoclasts in the blood. TRAP5B is increased in GD and correlates with patients who have osteoporosis. RANKL promotes osteogenesis, which is the formation of osteoclasts. RANKL was noted to be elevated in GD with correlation to osteopenia.
NCT04055831
Real-world effectiveness and safety of velaglucerase alfa in pediatric patients with Gaucher disease
By Andrew Friedman
Gaucher disease (GD) is a lysosomal storage disorder that is caused by an inherited deficiency or absence of the enzyme glucocerebrosidase. Of the three main disease types in GD, types 1 and 3 can have similar somatic presentation (e.g., growth retardation, thrombocytopenia, splenomegaly, bone crises), but type 3 is also associated with neurological symptoms (e.g., ataxia, slow horizontal saccades, seizures).
Velaglucerase alfa is indicated as enzyme replacement therapy (ERT) for patients with type 1 Gaucher disease, but more data is needed for those younger than four years of age.
This phase 4, observational, retrospective/prospective, non-controlled, non-comparative, single-center, real-world study (NCT04721366), will investigate the effectiveness of velaglucerase alfa on growth and symptoms as standard of care (SOC) in neonatal and pediatric patients with a confirmed diagnosis of type 1 or type 3 GD. Eligible participants will be patients who, aged ≤4 years, started treatment with intravenous velaglucerase alfa with 60 Units/Kg of intravenous velaglucerase alfa every other week. Depending on the patient’s age at enrollment, sufficient data will be collected retrospectively up to 36 months and prospectively up to 18 months to cover a total treatment period of 18–36 months from ERT initiation. Primary outcomes such as hemoglobin level, platelet count, and both liver and spleen volumes often change from ERT, especially for patients of up to five years of age. Additionally, a proportion of patients with growth normalization often show changes in their bone disease and thrombocytopenia after being treated with ERT. The secondary outcome is the number of adverse events. Plasma samples collected per SOC will be assayed for GD biomarkers, including glucosylsphingosine (lyso-GL1).
The study began in January 2021, and as of January 2022, LDRTC has enrolled nine patients (target, n=20). The anticipated time of completion is January 2023. The study findings will inform the case for earlier diagnosis and ERT initiation in GD, potentially improving clinical management of the disease.
Goker-Alpan et al, 101 World Symposium 2022

The 7th Genetic Rare and Immune Disorders Symposium (GRIDS) was held virtually on the 1st of July 2021.
LDRTC hosted physicians and researchers from the US and worldwide to address concepts, challenges, and new directions in the era of personalized medicine related to lysosomal storage disorders.
They discussed molecular and cellular aspects of clinical diversity and disease progression during the event. Another researcher discoursed about the next generation of molecular investigations in Lysosomal diseases.
Ozlem Goker-Alpan, MD, closed the first session by speaking about the integration of patient aspects with disease mechanisms for patient-centric care.
The summit also presented preclinical studies and clinical trials towards individualized therapies. Efficacy and safety in studies using gene therapy exposed promising clinical results were also highlighted.
Researchers explained how to target autophagy to overcome lysosomal storage disorders (LSDs) and pharmacological chaperones used to treat lysosomal storage disorders.
In the second session of the event, experts addressed novel technologies and emerging therapies for precision medicine, followed by digital health tools and services used to improve the treatment of patients.
They also highlighted pharmacological small molecules for the treatment of lysosomal storage disorders and how to use gene therapy approaches to address emerging challenges.
The last topic presented a patient-centered approach that guides treatment decisions in patients with rare diseases.

Inflammatory pathways and cardiac growth factors associated with Fabry disease cardiomyopathy
In Fabry disease, cardiovascular complications are most frequently encountered, contributing substantially not only to morbidity but also are the leading cause of premature death in both male and female patients with FD.
By Rekha Gopal
Fabry disease (FD) is an X-linked disorder where mutations in the GLA gene result in a deficiency of the enzyme ɑ-galactosidase A (ɑ–Gal A). There is a subgroup of FD patients with significant residual ɑ–Gal A activity having a phenotype with primary cardiac involvement occasionally referred to as “cardiac variant.”
In Fabry patients, cardiovascular complications are most frequently encountered, contributing substantially not only to morbidity but also are the leading cause of premature death.
The manifestations of cardiac involvement in Fabry disease are left ventricular hypertrophy (LVH), diastolic dysfunction, and microvascular angina. However, hypertrophic cardiomyopathy (HCM) is the most common cardiac pathology and cause of death in patients with FD.
The primary objective of this study is to identify blood-based biomarkers such as Gb3 and Lyso Gb3 for early detection of cardiac involvement and identification of different stages of HCM pathology in patients with Fabry disease. The accumulation of these substances within the myocardium releases inflammatory molecules and transforms growth factors that affect cardiac function, resulting in progressive cardiovascular diseases in FD patients.
The specific aim of this study is to assess the correlation between inflammatory markers in peripheral blood with the different HCM stages in FD and to analyze the association between inflammatory markers, growth factors, Lyso-Gb3 accumulation, and cardiac involvement in Fabry disease patients. This study also evaluates the role of the time of initiation and duration of therapy (ERT), respectively, in the activation and reversibility of inflammation-growth factor cascade and how these relate to the specific HCM stages in patients with FD.
The study involves 50 patients with 3 cohorts: 1. Fabry disease subjects with cardiomyopathy (20). 2. FD subjects without cardiomyopathy (20). 3. non-FD age matched controls with no known cardiac disease (10) (healthy controls).
The study will take place over an 18-month (or two-year) period. Biomarkers potentially related to cardio pathology will be measured in the Fabry disease patients and compared to that of the healthy controls.
The patients will be followed for a total of 18 months, during which blood and urine will be collected at 6-month intervals. The study began in November 2020. The first subject was recruited in December 2020, and the last subject signed consent in November 2021.
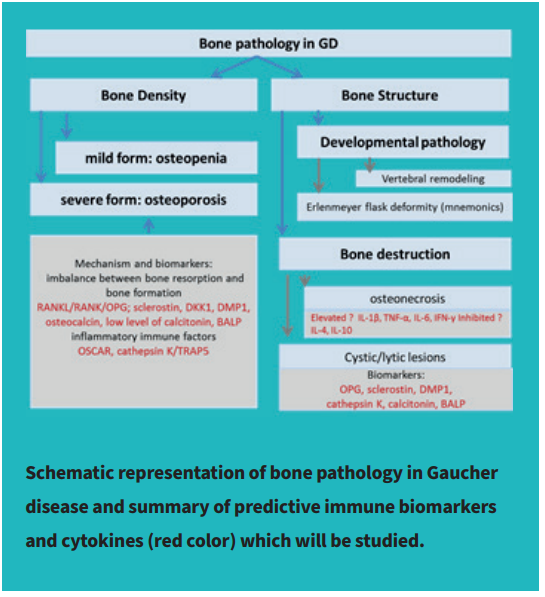
A plasma biomarker for osteopenia and osteoporosis in Gaucher disease Bone pathology in Gaucher disease introduction
By Margarita M. Ivanova, PhD, Ozlem Goker-Alpan, MD, Julia Dao, and Lauren Noll
The progressive bone disease occurs in 75% of patients with type 1 GD. One of Gaucher disease’s early signs involving bone pathology is the “Erlenmeyer flask” deformity that affects long bones and abnormality of bone modeling. Later, the majority of GD patients develop skeletal complications, including osteopenia and osteoporosis. Gaucher disease patients with skeletal involvement could be asymptomatic or present with symptoms such as pain, pathological fractures, cystic changes, or osteonecrosis.
The GD bone pathology is the result of the alterations in osteoclasts function and osteoblasts participating in bone remodeling and osteoclast differentiation due to extensive inflammatory response against toxic metabolite glucosylsphingosine (Lyso-Gb1). Reduced bone density leads to progressive osteopenia, osteoporosis. In addition, the aberrations in bone structure lead to abnormal vertebral remodeling and bone modeling, including Erlenmeyer flask deformity. Other bone structural abnormalities in Gaucher disease include osteonecrosis and lytic lesions.


TRAP5B APPLICATION AND DIAGNOSTIC RELEVANCE DESIGN:
TRAP5B APPLICATION IN GAUCHER DISEASE:
Interestingly, total TRAP, not TRAP5b, has been used as a biomarker for GD along with angiotensin converting enzyme (ACE), CHITO, and ferritin, all of which are markers for activated macrophages. However, the TRAP test presents a mixed level of two biomarkers: TRAP5a and TRAP5b. Recently, our research team demonstrated that elevated TRAP5b level correlates with osteopenia and osteoporosis progression in GD. Thus, enhanced TRAP5b level confirms the osteoclasts activation in GD patients. Moreover, plasma TRAP5b positively correlates with clinical biomarkers of GD pathology: CCL18, Lyso-Gb1, and chitotriosidase. Therefore, a possible mechanism underlying the activation expression of TRAP5 in osteoclasts is Gb1 accumulation in bone marrow cells, including monocytes/macrophages precursors cells and osteoclasts.
J Clin Med 2021 May 20;10(10):2217. doi:10.3390/jcm10102217
Ambroxol decreases heparan sulfate levels and improves lysosomal functions in vitro in MPS III patient-derived primary cell lines
By Margarita M. Ivanova, PhD, Ozlem Goker-Alpan, MD, and Julia Dao
Ambroxol is an anti-inflammatory and a Na+ channel blocker that has been used in the treatment of bronchopulmonary diseases. It was identified as a pharmacological chaperone that enhances the lysosomal enzyme activity, glucocerebrosidase, whose deficiency causes Gaucher disease (GD).
Sanfilippo syndrome or Mucopolysaccharidosis type III (MPS III) is a group of disorders caused by the deficiency of several enzymes involved in lysosomal degradation of heparan sulfate (HS). The MPS III clinical subtypes (A, B, C, and D) are each caused by the deficiency of enzymes. This condition leads to the primary Central Nervous System (CNS) involvement, presenting initially with neuropsychiatric symptoms progressive loss of cognitive skills. In the later stages of the disease, patients develop seizures, swallowing and feeding difficulties, progressive loss of mobility, and dementia. While gene therapy is currently being evaluated in clinical trials for MPS III patients who are not yet symptomatic or at an early disease stage, there is no established treatment for MPS III patients. Enzyme replacement therapy is not accessible to CNS and has not been proven to be effective in MPS III. Small molecules have a potential for CNS access, for example, pharmacologic chaperone therapies.
Ambroxol is an anti-inflammatory and a Na+ channel blocker that has been used in the treatment of bronchopulmonary diseases. Additionally, it was identified as a pharmacological chaperone that enhances the lysosomal enzyme activity, glucocerebrosidase, whose deficiency causes Gaucher disease (GD). Recent studies in GD and Parkinson’s diseases indicate that ambroxol helps to reduce the unwanted build-up of the toxic protein or substrates.
Our center investigates the effects of ambroxol activity in terms of alterations in heparan sulfate accumulation and lysosomal function in peripheral blood mononuclear cells (PBMC) derived from MPS III-A, MPS III-B, MPS III-C, and MPS III-D patients. No toxicity was observed in PBMC with concentrations of 1 and 10 µM ambroxol. Ambroxol activates the fusion of autophagic vesicles with lysosomes in PBMC and increases lysosomal acidification. Additionally, ambroxol treatment decreases heparan sulfate levels in PBMC in vitro.
Lysosomal dysfunction due to heparan sulfate accumulation may have wide-reaching consequences in MPS III pathology. Our data suggest that ambroxol may function as a therapeutic agent for patients with MPS III by acting as an autophagy-lysosomal enhancer. Further clinical studies are underway to assess the clinical utility of ambroxol in patients with MPS III.
https://www.mps2021.com
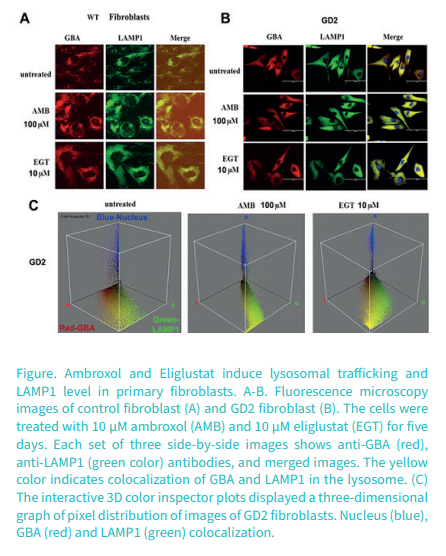
Cellular and biochemical response to chaperone versus substrate reduction therapies in neuropathic Gaucher disease
By Margarita M. Ivanova, PhD, Ozlem Goker-Alpan, MD, and Julia Dao
Gaucher disease (GD) is caused by a deficiency of the lysosomal membrane enzyme glucocerebrosidase (GCase) and the subsequent accumulation of its substrate, glucosylceramide (GC). Mostly, missense mutations of the glucocerebrosidase gene (GBA) cause GCase misfolding, inhibiting proper lysosomal trafficking, and impairs the autophagy pathway.
GD types 2 and 3 (GD2-3), or the neuronopathic forms, affect the Central Nervous System (CNS) and have severe systemic involvement and progressive bone disease. Enzyme replacement therapy (ERT) is the standard of care in GD for the treatment of systemic symptoms: splenomegaly, hepatomegaly, thrombocytopenia, and low platelets. Unfortunately, ERT is not effective in treating CNS pathology because of a lack of access through the Blood-Brain Barrier (BBB). Other alternative therapy modes to access CNS are using small molecules that may cross the BBB, such as new generation forms of substrate reduction therapy (SRT) and pharmacologic chaperones (PCT).
Ambroxol (AMB) is a pharmacologic chaperone that partially recovers the mutated GCase activity and could potentially treat the neurological symptoms due to crossing the blood-brain barrier.
Eliglustat (EGT), an inhibitor of UDP-glucosylceramide synthase, catalyzes GC biosynthesis, reducing GC influx load into the lysosome. Substrate reduction therapy (SRT) using EGT is associated with improvement in GD bone marrow burden score and bone mineral density paralleled with the improvement in hematological parameters.
Our research group assessed the effects of EGT and AMB on GCase enzyme activity and autophagy-lysosomal pathway (ALP) in primary cell lines derived from patients with GD2-3. We found that EGT, same as AMB, enhanced GCase activity in control cells and that an individualized response that varied with GBA mutations was observed in cells from patients with GD2-3. EGT and AMB enhanced the formation of lysosomal/late endosomal compartments and improved autophagy, independent of GBA mutations. Both AMB and EGT increased mitochondrial mass and density, suggesting enhancement of mitochondrial function by activating the mitochondrial membrane potential. These results demonstrate that EGT and AMB, with different molecular mechanisms of action, enhance GCase activity and improve autophagy-lysosome dynamics and mitochondrial functions.
PLoS One 2021 Oct 25;16(10):e0247211. doi: 10.1371/journal.pone.0247211.
CME Series
LDRTC co-hosted four webinars as part of the CME series. In these webinars Dr. Goker-Alpan, and a few top LSD experts addressed the latest research and treatment regarding lysosomal storage diseases (LSDs).
The first module addresses current and emerging therapies for lysosomal storage disease, followed by new insights into lysosomal storage diseases, and how pathophysiology is changing treatment. The program also presents new and emerging phenotypes in lysosomal storage diseases and central symptoms and comorbidities regarding LSDs.
Ozlem Goker-Alpan, MD, and Ari Zimran, MD, open the series introducing current and emerging therapies for lysosomal storage diseases. Enzyme replacement therapy (ERT), substrate reduction therapy (SRT), hematopoietic stem cell transplantation, and gene therapy (in-vivo and ex-vivo) are treatments available for LSDs patients. They also present emerging therapies, including new SRTs, and Arimoclomol. According to Dr. Zimran, to maximize treatment success, patients must start treatment before the development of irreversible complications.
In this second webinar, Ozlem GokerAlpan, MD, and Gregory Grabowski, MD, explore the pathophysiology of lysosomal storage diseases and how new research may improve patient management. Dr. Goker-Alpan starts this module by highlighting the importance of precision (personalized) medicine. This tailored approach helps selecting optimal therapies based on a patient’s genetic, molecular, or cellular analysis. An in-depth understanding of the pathophysiology of disorders like Gaucher disease, Fabry disease, Sanfilippo syndrome, etc, helps physicians to better treat their patients.
In the third activity, Uma Ramaswami, MD, and Ozlem Goker-Alpan, MD, describe the traditional phenotypes for lysosomal storage diseases. They discourse about emerging phenotypes in LSD patients after receiving new treatments. Dr. Ramaswami highlights the importance of a multidisciplinary approach when treating patients with LSDs.
The fourth webinar closes the CME series with doctors Ozlem Goker-Alpan and Swati Sathe addressing central symptoms and comorbidities related to lysosomal storage disorders. Together, they unveil LSDs with significant psychological/behavioral symptoms. Dr. Sathe presents correlations between lysosomal storage disorders and diseases such as Parkinson’s disease or depression. She also explains the most appropriate methods to monitor and measure neurological symptoms in LSDs. Dr. Sathe ends her presentation by showing how treatment impacts neurological symptoms in certain LSDs. Watch the webinars at https://checkrare.com

Lysosomal Storage Disease Quarterly CME/CE Webinar Series
Program Description:
Quarterly CME Series on Lysosomal Storage Disease (LSD), developed and presented by leading LSD faculty from around the world; hosted by Dr. Goker-Alpan, Founder and CMO of the Lysosomal and Rare Disorders Research and Treatment Center (LDRTC); and provided through collaboration between CheckRare, a leading publisher and learning platform focused on rare diseases and AffinityCE, an accredited medical education company with a rare disease focus and program experience.


