

Letter from the Director
Ozlem Goker-Alpan, MD
Drug Development as a Business Decision Impacts Patients with Rare Disorders
Last week, I got the news that a trial that might offer the only hope for an otherwise deadly disorder in infants will not be pursued. Within the complex and evolving landscape of healthcare, drug development represents a critical intersection of science, business, and ethics. Bringing new drugs to market is a process often driven by potential profitability. This business-centric approach significantly influences the development of treatments for rare disorders where the patient population is small and the return on investment is often uncertain. Patients with rare disorders and their clinicians often find themselves at a complex and frustrating crossroads. The high cost of drugs and the pharmaceutical industry’s hesitancy to pursue drug development for less profitable conditions significantly impact the quality of life and treatment options for these patients.
Pharmaceutical companies, like all businesses, aim to maximize profits. Drugs for common conditions such as diabetes, hypertension, and cholesterol management have a guaranteed market, ensuring steady revenue streams. In contrast, treatments for rare disorders – known as orphan drugs – serve a small number of patients, making them less commercially attractive despite their often higher pricing. Developing a new drug is a costly and risky endeavor. The process from discovery through clinical trials to market approval can take over a decade and cost upwards of over $2 billion. This high investment in terms of time and finance naturally steers pharmaceutical companies towards drugs that promise high returns, typically those targeting common diseases with large patient populations.
Clinicians are often caught in the ethical dilemma of wanting to provide the best possible care but are constrained by the realities of the healthcare system. They witness the disparities in treatment access and are forced to navigate these inequities on a daily basis, which can be distressing and demoralizing.
The decision by pharmaceutical companies not to pursue drug development for certain rare disorders due to low return on investment leaves patients feeling neglected and marginalized. In addition, the high cost of already existing drugs and the selective nature of drug development based on profitability disenfranchise patients with rare disorders, leaving them and their clinicians in a precarious position. Patients face financial burdens, limited treatment options, and the emotional toll of feeling forgotten by the system. Addressing these issues requires a concerted effort from governments, pharmaceutical companies, healthcare providers, and patient advocacy groups to ensure that profitability does not overshadow the fundamental right to health and equitable treatment access.
The orphan drug legislation, offering incentives like tax credits, grants for clinical research, market exclusivity, and regulatory fee waivers, may aim to offset the lower profitability of orphan drugs and encourage their development. While this has led to an increase in orphan drug approvals, the question of accessibility and affordability remains critical. Orphan drugs, once developed, are often exorbitantly priced, reflecting the high cost of their development and the need to recoup investments from a smaller patient base. This pricing model can make these drugs inaccessible to many patients, especially in countries without robust healthcare funding or insurance coverage for rare diseases. Given that none of the rare disorders pose a real public health concern, in developed countries such as the US and UK, the clinicians are utilized as gatekeepers of medical economics and are sometimes expected to put the financial burden of patient care in front of what is the best interest of the patient. I am mostly reminded by the cost of the drugs that I prescribe by the “Managed Care” companies that actually manage the care of my patients without knowledge or expertise in that specific rare disorder.
Patient advocacy groups play a crucial role in highlighting the needs of those with rare disorders. Collaborations between these groups, pharmaceutical companies, and government bodies can lead to more effective strategies for drug development and funding. Such partnerships can also ensure that the voices and needs of patients are heard in the drug development process. The development of Kalydeco, in collaboration with 65 Roses, is a prime example of how patient advocacy groups can play a crucial role in drug development. This partnership not only led to a groundbreaking treatment for CF but also set a precedent for how pharmaceutical companies and patient organizations can work together to accelerate medical advancements. 65 Roses, a patient advocacy group named for the way some children mispronounce “Cystic Fibrosis,” played a pivotal role in the development of Kalydeco. This collaboration highlighted several key aspects: 65 Roses provided invaluable insights into the patient experience of living with CF. This perspective was crucial in shaping the research and development process. In addition, patient advocacy groups, including 65 Roses, have long been instrumental in raising funds for CF research. Their contributions were vital in supporting the early stages of Kalydeco’s development. The success of Kalydeco underscores the importance of patient-centric approaches in developing treatments for complex genetic diseases.
Drug development, inherently a business decision, has profound implications for patients with rare disorders. While profitability drives the pharmaceutical industry, the need for treatments in rare diseases presents both a challenge and an opportunity for a more inclusive approach that will engage both the patients and clinicians who are not only experts in the field but also have mutual devotion and responsibility for these patients. Balancing business objectives with ethical responsibilities requires innovative strategies, collaborations, and a commitment to ensuring that all patients, regardless of the prevalence of their condition, have access to effective treatments. The future of drug development for rare disorders lies in finding synergies between commercial success and the overarching goal of improving patient health and quality of life.

2023: A Year in Review Highlights from the LDRTC Clinical Trials and Research Unit
Lauren Noll
LDRTC ENROLLED OVER TWENTY PATIENTS ACROSS BOTH DISEASE GROUPS WHOSE DATA CONTRIBUTED TO FDA APPROVALS.

Throughout 2023, we successfully maintained and updated data for over two hundred Gaucher, Fabry, and Pompe patients across various registries. Approximately fifty patients throughout the year have been receiving regular treatment they wouldn’t have been able to receive commercially spread across numerous studies. We opened seven new studies, which consisted of one observational, two prescreening, one oral treatment, one infusion, and two gene therapy. Of those seven new studies, our site enrolled the first patient in four of them.
Our team attended various patient conferences across the country, including the Fabry Patient Conference in Greensboro, NC, Niemann-Pick in Orlando, FL, Gaucher Patient Conference in NJ, Sanfillipo in Hershey Park, PA, MPS in MD, and Tay-Sachs in Reston, VA. These patient conferences allow us to showcase our facility and what we can offer patients, as well as learning about new treatments and/or issues currently facing those who live with these rare genetic diseases.
The highlight of our year was seeing two treatments get approved by the FDA for commercial use. The first approval came in May 2023 for the treatment of Fabry disease. The Elfabrio (pegunigalsidase alfa) studies started in our office back in 2013 as Phase 1 studies, which were first in human. This medication is an enzyme replacement therapy (ERT), which is administered every two weeks. Since the initiation in 2013, we have run multiple Elfabrio studies, which include Phase 2, Phase 3, and expanded access with no gaps over the past ten years.
The second approval came in October 2023 for Pompe disease. Pombiliti (cipaglucosidase alfa-atga) and Opfolda (miglustat) are a combination therapy which consists of an ERT infusion and an oral chaperone every two weeks.
These combination therapy studies started in 2016 and have been running with patients enrolled since the start. We enrolled over twenty patients across both disease groups whose data contributed to these FDA approvals. The personal sense of pride that is felt knowing that our hard work directly contributed to new medications being approved by the FDA to treat these rare genetic diseases is hard to describe. We thank all of our patients who have been with us for years enrolled in these studies. It is because of you that we get to be a part of these major events that provide options for treatment for patients whom are affected by these diseases.
As we move through 2024, we plan to attend many patient conferences to continue our outreach to help positively impact the rare disease community. We are excited for the currently running studies, old and new, as well as the upcoming studies not yet open. Although we do not expect any FDA approvals for studies in which we have participated in this next year, we are seeing an increase in gene therapy studies coming down the pipeline. Gene therapy offers a level of excitement all of its own, given the potential impact this type of treatment will have for our patients. We look forward to continuing to be able to contribute to the advancement of new therapies for Lysosomal disorders through clinical trials research.

Revolutionizing Kidney Health in Patients with Fabry Disease: Unraveling Novel Urine Biomarkers for Early Detection of Nephropathy
Margarita Ivanova, PhD and Andrew Friedman
Traditional laboratory tests used to assess kidney function, such as serum creatinine and estimated glomerular filtration rate (eGFR), are not sensitive enough to detect early renal involvement in FD. At an early stage, clinical laboratory signs of kidney involvement, such as a decline in renal function, may be absent. However, the accumulation of Gb3 and Lyso-Gb3 can lead to inflammation and progressive podocyte loss in the kidneys. Podocyte loss is an irreversible event that impairs the glomerular filtration barrier, resulting in decreased kidney function. Inflammatory factors arising from kidney injury can serve as potential diagnostic and therapeutic biomarkers, offering valuable insights beyond current laboratory tests.
There is a growing interest in the exploration of urinary biomarkers as valuable tools for both the diagnosis and monitoring of nephropathy in FD. Urinary biomarkers hold promise as non-invasive tools in providing insights into the FD-related kidney disease. These biomarkers may include specific proteins, cellular elements, or metabolites that are excreted in urine and can be quantified to assess renal health. They offer several advantages, including non-invasive sample collection, potential early detection of renal involvement, and the ability to track disease progression over time. Gb3, the precursor molecule of Lyso-Gb3, accumulates in various tissues and organs of FD patients. While Lyso-Gb3 is a breakdown product of Gb3, the measurement of Gb3 itself can provide insights into the extent of glycolipid accumulation. In Fabry nephropathy, the kidneys are one of the primary sites of Gb3 accumulation. Quantifying urinary Gb3 levels can help assess renal involvement and guide the management of kidney-related complications. Gb3 measurements in urine can complement the assessment of Lyso-Gb3 levels, offering a more comprehensive view of glycolipid storage in the body.
While the primary pathological hallmark of FD is sphingolipid deposition, recent research has shown downstream events play a major role in disease progression and severity, that immune-based biomarkers in urine can provide valuable insights to further the understanding of the disease process. FD involves chronic inflammation, and the accumulation of Gb3 and its derivatives can trigger an inflammatory response in affected tissues, contributing to organ damage, including renal and cardiovascular complications. Excessively stored glycosphingolipids in cells can activate immune cells, leading to the release of pro-inflammatory cytokines. These cytokines play a role in initiating and sustaining the inflammatory response.

TGF-β1 and VEGF-A as Key Molecules for Advancing Cardiac Diagnostics in Fabry Disease
Margarita Ivanova, PhD
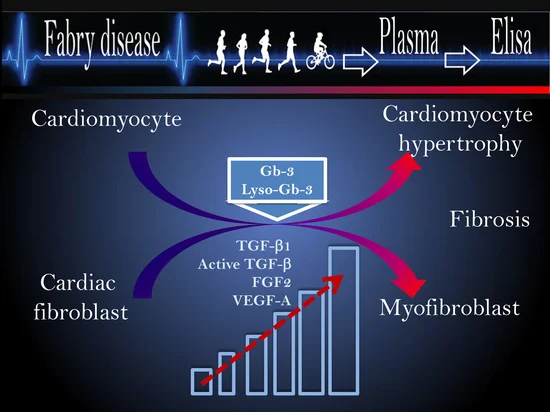
Fabry disease (FD) is a Lysosomal disorder that occurs due to α-galactosidase A deficiency, causing the accumulation of globotriaosylceramide (Gb3) and its metabolite globotriaosylsphingosine (Lyso-Gb3). While FD occurs in a spectrum of manifestations, the most common symptoms of FD include cardiovascular complications and hypertrophic cardiomyopathy (HCM). Cardiovascular complications contribute substantially to morbidity in FD and are the leading cause of premature death in male and female patients.
Clinical diagnostic tools such as echocardiogram and cardiac MRI are used to evaluate cardiac involvement, but due to variations in hypertrophic cardiomyopathy and myocardial fibrosis patterns in patients with FD, it is essential to identify biomarkers that can predict early cardiac outcomes.
In our center, we studied whether levels of circulated bloodstream growth factors, such as TGF-β1 and VEGF-A, could be used to diagnose and monitor cardiac damage progression in patients with Fabry disease. This research has the potential to improve the accuracy and efficiency of diagnosing and treating Fabry disease-associated cardiomyopathy, ultimately leading to improved outcomes for patients.
We found that the elevated TGF-β1 correlates with HCM and myocardial fibrosis in male and female FD patients, indicating its potential biomarker for diagnosis of an early stage of cardiac fibrosis, even before hypertrophy is detected. Moreover, the elevation of TGF-β1 and active-TGF-β1 associated with Lyso-Gb3 elevation provides evidence of a chronic inflammatory state and the activation of fibrosis in FD patients [1].
Angiogenesis biomarker VEGF-A correlates with plasma Lyso-Gb3 and is associated with hypertrophic cardiomyopathy in FD patients. Thus, serum TGF-β1 and VEGF are predictive biomarkers for adverse cardiovascular events in FD.
Gender differences in the secretion of TGF-β1, VEGF, and FGF2 can explain patterns of cardiac involvement in male vs. female FD patients, with fibrosis occurring early in the course in females.

2. Frangogiannis NG. Transforming growth factor-ꞵ in myocardial disease. Nature Reviews Cardiology. 2022;19(7):435-55. doi: 10.1038/s41569-021-00646-w.
3. Braile M, Marcella S, Cristinziano L, Galdiero MR, Modestino L, Ferrara AL, et al. VEGF-A in Cardiomyocytes and Heart Diseases. International Journal of Molecular
Sciences. 2020;21(15):5294. PubMed PMID: doi:10.3390/ijms21155294.
Biomarkers for Detection and Monitoring of Cardiac Damage in Fabry Disease
Ozlem Goker-Alpan, MD
These biomarkers provide the objective tools to diagnose, assess disease severity, monitor progression, and guide therapeutic interventions.
While FD manifests in various ways, its most common symptoms include cardiovascular complications and hypertrophic cardiomyopathy (HCM). These cardiovascular issues significantly contribute to morbidity and, unfortunately, serve as the primary cause of premature death in both male and female FD patients.
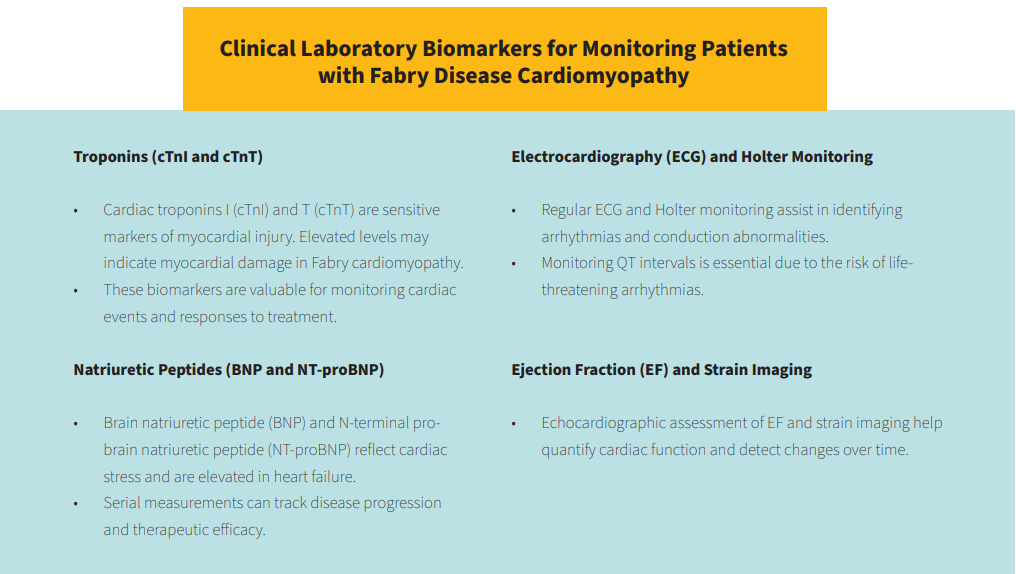
Timely diagnosis and effective management of Fabry disease cardiomyopathy are critical for improving patient outcomes. Clinical biomarkers play a pivotal role in this regard, offering valuable insights into the cardiac status of affected individuals. These biomarkers provide the objective tools to diagnose, assess disease severity, monitor progression, and guide therapeutic interventions.
Biomarkers play a crucial role in the diagnosis and monitoring cardiomyopathy in Fabry disease. Early detection of the disease, assessment of cardiac involvement, and tracking disease progression are essential for effective management and improving patient outcomes.
There are clinical tools, such as echocardiograms and cardiac MRI scans, that are employed to assess cardiac involvement. However, the variations in hypertrophic cardiomyopathy and myocardial fibrosis patterns among FD patients highlight the need for biomarkers that can predict early cardiac outcomes.
We have recently undertaken a study to explore the potential of immune markers, particularly TGF-β1, known as the master regulator of fibrosis, and VEGF-A, a key mediator of angiogenesis, in evaluating cardiac manifestations in Fabry disease (FD).
Our current research at the Lysosomal Disease Treatment and Research Center (LDRTC) has uncovered that heightened levels of TGF-β1 are associated with the initial stages of cardiomyopathy (HCM) as well as myocardial fibrosis in FD patients across genders.
This suggests that TGF-β1 could serve as an early biomarker for cardiac fibrosis, detectable even before the onset of hypertrophic changes. Additionally, the concurrent elevation of TGF-β1 and its active form alongside increased Lyso-Gb3 levels points to a sustained inflammatory response and the initiation of fibrosis in FD patients.
This inflammatory state is further characterized by the dysregulation of inflammatory cytokines, which contribute to the pathogenesis and progression of cardiac involvement in FD, marking a potential therapeutic target for early intervention [1].
2. Frangogiannis NG. Transforming growth factor-ꞵ in myocardial disease. Nature Reviews Cardiology. 2022;19(7):435-55. doi: 10.1038/s41569-021-00646-w.
3. Braile M, Marcella S, Cristinziano L, Galdiero MR, Modestino L, Ferrara AL, et al. VEGF-A in Cardiomyocytes and Heart Diseases. International Journal of Molecular
Sciences. 2020;21(15):5294. PubMed PMID: doi:10.3390/ijms21155294.
The First 3D Bioprinted In-Vitro Bone Model for Gaucher Disease
Margarita Ivanova, PhD
GD primarily affects monocyte lineage cells, including macrophages, which play essential roles in the immune system and are involved in osteoclast differentiation and osteoclast–osteoblast communication during bone development and remodeling. 80% to 95% of patients with GD, including asymptomatic ones, present with varying forms of bone involvement, such as structural changes, severe bone pain, and osteoporosis.Alteration of bone remodeling in GD causes various structural bone pathologies such as Erlenmeyer flask deformity, bone modeling abnormality, osteonecrosis, lytic lesions, and early osteoporosis. However, our understanding of the cellular aspects involved in abnormal bone remodeling is still incomplete.
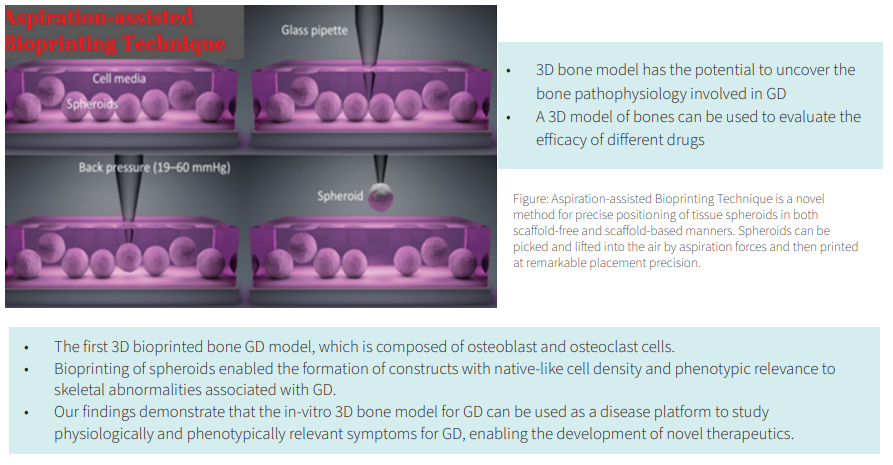
The translational research group from our center collaborated with Dr. Ozbolat from Penn State University to develop the first 3D human model of GD using aspiration-assisted freeform bioprinting. This technology provides a platform device for decoding the cellular basis of developmental bone abnormalities in GD.
The techniques include the formation of spheroids from co-cultured human bone marrow-derived mesenchymal stem cells and peripheral blood mononuclear cells derived from GD patients, followed by differentiation into osteoblast and osteoclast lineages.
Co-differentiated spheroids were then 3D bioprinted into rectangular tissue patches that mimic bone tissue.
The results revealed positive alkaline phosphatase (ALP) and tartrate-resistant ALP activities, with multi-nucleated cells demonstrating the model’s efficacy, corroborating with gene expression studies. There were no significant changes in differentiation to osteogenic cells but pronounced morphological deformities in spheroid formation, more evident in the ‘severe’ cohort.
Overall, the presented GD model has the potential to be adapted to personalized medicine not only for understanding the GD pathophysiology but also for personalized drug screening and development.
Deciphering sanfilippo syndrome: Insights and Prospects for Promising Adjunct Therapies
Arooj Agha

The most severe subtype, Type A (MPS IIIA), is caused by a deficiency in the enzyme sulfamidase or heparan N-sulfatase. Patients with Type A experience rapid symptom progression, including developmental delays, progressive intellectual disabilities, hyperactivity, speech impairment, and coarse facial features. Type B (MPS IIIB) is characterized by a deficiency in the enzyme α-N-acetylglucosaminidase, and patients exhibit similar symptoms to Type A, such as intellectual disability, developmental delays, and speech problems. Type C (MPS IIIC) involves a deficiency in the enzyme heparan acetyl CoA: α-glucosaminide N-acetyltransferase, and symptoms progress more slowly compared to Types A and B. Patients may experience developmental delays and behavioral issues like hyperactivity. The rarest subtype, Type D, results from insufficient N-acetylglucosamine 6-sulfatase production and activity, progressing faster than Type C but similar to Types A and B. Since MPS III is a neurodegenerative disease, neuropathic manifestations are expected in each subtype.
The “Natural History Study of Patients with Sanfilippo Disease(s) (MPS3)” aims to collect unique biomarker data for all types of the disease over a 6-month period, with at least three specimen collection time points. The monitored biomarkers include serum heparan sulfate, urinary glycosaminoglycans (GAGs), and serum neurofilament light chain (NfL), which is a promising biomarker for tracking neurological symptom progression. This study aims to establish baseline trends for each biomarker and track changes over time. After collecting and analyzing this initial data, a follow-up treatment study will be initiated, administering Ambroxol to enrolled patients. The same biomarkers will be collected during the treatment phase for consistency and to monitor biomarker trends and changes.
Decoding Bone Pathology in Pediatric Gaucher Disease Through Exploration of Immune Biomarkers and Growth Factors
Heather Goodwin

Bone disease associated with GD results from a complex set of accumulated events. These include abnormal bone development presenting as a vertebral remodeling defect and abnormalities in the growth process of long bones.
A hallmark of GD on radiological imaging is the Erlenmeyer flask deformity (1, 2). Bone destruction can occur due to osteonecrosis, and cystic/lytic lesions may be observed with or without avascular necrosis. Reduced bone mineral density can lead to pathologic fractures, which can start in teenage years and result in early osteoporosis in both female and male patients with GD.
Pain is one of GD’s prime and debilitating symptoms, often associated with other structural skeletal involvement (3).
Normal bone and skeletal growth is influenced by both environmental and genetic factors. During childhood, rapid bone growth occurs as bone tissue is added to the epiphyseal plate, which lengthens the bones. Bones are thickened as bone tissue is added to the surface, increasing the diameter. The process of bone remodeling and repair continues after birth and into adulthood. The underlying mechanisms of regulation and complications of bone development in pediatric GD patients, however, are not yet fully understood. Abnormalities of skeletal growth and bone turnover could be a result of abnormal regulation by growth factors. Given that chronic inflammation leads to alterations in the function and differentiation of osteoclasts and osteoblasts, which participate in bone growth and remodeling, and that a cascade of immune-mediated inflammatory reactions is set off by substrate deposition in GD, we hypothesize that these inflammatory events interfere with the normal process of bone growth, mineralization, and remodeling in pediatric GD patients.
To study this, we are recruiting ten GD patients aged five to twenty-one years with and without bone involvement to participate for a period of twelve months. Plasma samples from healthy children in the same age range will be used as controls. Skeletal involvement will be assessed using DEXA scans, X-rays, and patient-reported outcome surveys; biochemical data will be obtained from blood and urine samples. Combined, this data will allow us to evaluate the relationship of GD-mediated inflammation and growth factor dysregulation with bone involvement seen in pediatric GD patients.
Further, we aim to identify blood-based biomarkers correlated with bone involvement in GD patients to assess the correlation between levels of cytokines and other inflammatory markers with the severity of bone involvement, and to analyze the association of growth factors and Lyso-Gb1 (a biomarker of GD) with markers of macrophage activation and clinical parameters in pediatric GD patients.
This clinical study (ClinicalTrials.gov ID: NCT 06116071) commenced in December 2023 and will be actively enrolling patients until early 2025.

References |
1. Kaplan P, Andersson HC, Kacena KA, Yee JD. The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Arch Pediatr Adolesc Med. 2006;160(6):603-8.
2. Goker-Alpan O. Therapeutic approaches to bone pathology in Gaucher disease: past, present and future. Mol Genet Metab. 2011;104(4):438-47.
3. Devigili G, De Filippo M, Ciana G, Dardis A, Lettieri C, Rinaldo S, et al. Chronic pain in Gaucher disease: skeletal or neuropathic origin? Orphanet J Rare Dis. 2017;12(1):148.
Gaucher Disease and Immune Thrombocytopenic Purpura (ITP)
Lia Van, NP-C and Leah Svarny, PA-C

On examination, the patient looked pale and tired. No petechiae or purpura were noted. Spleen and liver were enlarged. The spleen was palpable approximately 3-5 cm below the costal margin, and the liver edge was palpable. Neurologically, the patient was at baseline.
Labs was ordered, including CBC w/diff and coagulation panel. From this sample, labs reviewed platelets to be 17k (ref.140-400K), Hgb 9.0 (ref. 11.7-15.5g/dL), and Hct: 28.9 (ref. 35.0- 45%). aPTT/PT were WNL and elevated D-dimer of 0.56 (ref. <0.5mcg/mL). Previous platelet level on June 9, 2023, was 139K, and 243K on January 1, 2022, both with normal Hgb and Hct. A confirmatory repeated CBC showed platelets: 22K, Hgb: 9.2, and WBC: 4.2 (ref. 4.5-13 thousand/uL). Patient was recommended to be evaluated by Children’s Hospital. There, a CT of the abdomen was done, showing an increase in spleen size of 17.4 cm from 15 cm in August 2023.
On November 17, 2023, there was a workup for thrombocytopenia/pancytopenia, including antiplatelet antibodies, viral titers, CMP 20, reticulocyte count, and haptoglobin. On this specimen, the platelet count was 18K, Hgb was 8.0, MCV 25.7 (ref. 34-46) WBC was 4.2 (ref. 4.5-13.0 thousand/uL). Smear showed nucleated RBC with microcytosis, poikilocytosis, ovalocytes, and hypochromasia. Total iron 13 (ref: 27-164 mcg/dL), TIBC normal. Antiplatelet antibodies (qualitative was positive for IgG and IgM). Patient was referred to pediatric hematology for evaluation and treatment. The patient was diagnosed with Immune Thrombocytopenia (ITP).
For treatment, she received a high dose of IVIG. After the 1st dose of IVIG, follow-up labs showed a platelet count of 87K, WBC count of 5.1, and Hgb of 9.5gm/dl. Reticulocyte count of 4.0 (0.5-1.5). Patient responded well with IVIG.

References | 1. Mistry PK, Sadan S, Yang R, Yee J, Yang M. Consequences of diagnostic delays in type 1 Gaucher disease: The need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am. J. Hematol. 2007;82:697–701. doi: 10.1002/ajh.20908. 2. Motta I, Consonni D, Stroppiano M, Benedetto C, Cassinerio E, Tappino B, Ranalli P, Borin L, Facchini L, Patriarca A, Barcellini W, Lanza F, Filocamo M, Cappellini MD; Splenomegaly Gaucher group. Predicting the probability of Gaucher disease in subjects with splenomegaly and thrombocytopenia. Sci Rep. 2021 Jan 28;11(1):2594. doi: 10.1038/s41598-021-82296-z. PMID: 33510429; PMCID: PMC7843616. 3. Weycker D, Hanau A, Hatfield M, Wu H, Sharma A, Bensink ME, Chandler D, Grossman A, Tarantino M. Primary immune thrombocytopenia in US clinical practice: incidence and healthcare burden in first 12 months following diagnosis. J Med Econ. 2020 Feb;23(2):184-192. doi: 10.1080/13696998.2019.1669329. Epub 2019 Oct 9. PMID: 31547724. 4. Izak M, Bussel JB. Management of thrombocytopenia. F1000Prime Rep. 2014 Jun 2;6:45. doi: 10.12703/P6-45. PMID: 24991422; PMCID: PMC4047949. 5. Immune thrombocytopenia (ITP) | NHLBI, NIH. (2022, March 24). NHLBI, NIH. https://www.nhlbi.nih.gov/health/immune-thrombocytopenia 6. Harvard Health. (2022, March 11). Immune thrombocytopenic purpura (ITP). https://www.health.harvard.edu/a_to_z/immune-thrombocytopenic-purpura-itp-a-to-z
Comprehensive CME Series on Lysosomal Disorders
Dr. Walla Al-Hertani opens the series by discussing building and maintaining a multidisciplinary team for Lysosomal disorders. Dr. Al-Hertani outlines challenges in the current care models for Lysosomal disorders and how precision medicine fits in.
The following lecture overviews managing cardiomyopathies in Lysosomal disorders.
John Jefferies, MD, describes the cardiologist’s role in the team approach to care and lists best practices to manage and treat cardiomyopathies in Lysosomal disorders.
Dr. Goker-Alpan closes the last webinar of the CME Series covering the pulmonary manifestations of Lysosomal diseases with John Bach, MD. He speaks about assessing, monitoring, and managing respiratory involvement in Lysosomal disorders and explains how to prevent respiratory failure and avoid resorting to tracheostomies.

Predicting Nephropathy Progression in Fabry Disease
In this pilot study, urinary biomarkers related to inflammation and renal injury while compared to standard laboratory measures of kidney function, are evaluated. The study will involve fifty subjects aged eighteen to eighty, divided into four cohorts:

In summary, immune-based biomarkers in urine offer a promising avenue for understanding the inflammatory aspects of Fabry disease. By quantifying these biomarkers, researchers and clinicians can gain insight into the immune response, inflammation severity, and potentially tailored treatment strategies to address both the metabolic and immune components of this complex genetic disorder.
References | NCT06065605
Empowering Families: LDRTC’s Inaugural Gaucher Educational Meeting for Affected Families and Children
During the meeting, we launched “Our Journey with Gaucher Disease,” a book published by LDRTC with stories written by patients and their families impacted by Gaucher disease.

The guest speaker, Ravi Kamath, MD, PhD, educated the audience about the evaluation and management of the skeletal manifestations of Gaucher disease in children and young adults.
Ozlem Goker-Alpan, MD, Founder and CMO of the Lysosomal & Rare Disorders Research & Treatment Center, closed the event by thanking the attendees and reinforcing LDRTC’s dedication to the rare community. “We are committed to advancing the understanding and treatment of this disease and are delighted to offer this educational meeting.”
In summary, immune-based biomarkers in urine offer a promising avenue for understanding the inflammatory aspects of Fabry disease. By quantifying these biomarkers, researchers and clinicians can gain insight into the immune response, inflammation severity, and potentially tailored treatment strategies to address both the metabolic and immune components of this complex genetic disorder.



LDRTC celebrated its 9th GRIDS by hosting over two hundred attendees and researchers, in person and virtually, from Brazil, Canada, Colombia, Egypt, Germany, India, Ireland, Israel, Italy, Japan, Mexico, Morocco, Nepal, Pakistan, Portugal, Russia, Spain, Turkey, the United States and the United Kingdom.
The keynote speaker, Dr. Konrad Sandhoff, opened the summit, sharing his journey as a chemist into the world of sphingolipids and sphingolipidoses. He also addressed the mechanisms and impact of secondary ganglioside and lipid deposition in Lysosomal disorders.
During the first day of the symposium, experts discussed basic mechanisms in Lysosomal disorders - phenotypic diversity and cell biology, biomarkers and other genetic approaches in LDs, current clinical challenges in Lysosomal disorders - newborn screening and LDs in the neonatal period, and the application of novel technologies in LDs - role of AI in Lysosomal disorders.
On the second day of the event, experts presented the next-generation therapies in LDs, including developing therapies for diseases caused by the deficiencies of Lysosomal sialidases, delivering CRISPR to the brain, and small molecule therapies for primary and secondary sphingolipidosis. The summit continues with lectures on current therapeutic challenges in Lysosomal disorders and who is the candidate for gene therapy, Intrathecal vs. Trojan Horse approach in treating neuropathic LDs, the timing of initiation of ERT, and disease outcomes for patients with MPS.
After the presentations, a group of LD experts went to LDRTC to participate in the GRIDS Clinic. After reviewing patient records and visiting with them, they advised patients with debilitating disorders. If you would like to watch the CME lectures from 2023 GRIDS, please visit https://www.gridssymposium.org/

