

Letter from the Director
Ozlem Goker-Alpan, MD
The SARS-CoV-2 pandemic certainly led to unprecedented times with change of life and plans for all. LDRTC entered into 2020 with big plans and as much enthusiasm and commitment to our patients and staff as ever.
We were scheduled to relocate into a new and centralized location that would integrate our patient care with clinical and translational research facilities.
We had planned to organize our annual GRIDs symposium, this time at an international location. But then 2020 brought forth unique challenges and complications, both unforeseen and unpredicted, that left no one and no organization untouched worldwide! LDRTC had to readjust the ideas and revamp the plans to serve our patient community with just as much dedication as always.
The COVID-19 pandemic represents an enormous world-wide challenge both medically and economically. This challenge further intensifies multiple folds for the Rare Disease Community, including patients with lysosomal storage disease (LSD) and their families. Their medical care is not seen by many in the establishment as a primary concern during such large scale public health disasters.
Rare disease patients from all ages belong to the most vulnerable population not only because of their susceptibility to the catastrophic complications of the COVID-19 infection but also their need for continued access to high-level and integrated medical care. Their necessity of life-saving but expensive therapeutics or participation in ongoing clinical trials is hampered during the COVID-19 outbreak.
While social distancing and stay-at-home orders have been issued as a method to slow the curve of COVID-19 infections, it presents the rare disease community with its own set of unique complications. Enzyme replacement therapy (ERT), one of the most accepted treatment forms for various LSDs, requires periodic visits to hospitals or infusion clinics for treatment and medical evaluation by their care providers.
This pandemic has accelerated many healthcare technologies. We have been using Telemedicine to reduce our patient’s exposure to the virus while we guarantee their continued care. The novel research on COVID-19 has helped us with insights into the pathogenic mechanisms of lysosomal pathways related to SARS-CoV-2 infection that also exist in lysosomal disorders.
Inside this newsletter are some of the highpoints from LDRTC, that include our relocation to a new facility, new safeguards, and accommodations put in place to ensure the safety of patients under our clinical care and those participating in clinical trials, restructuring of GRIDS 2020, and highlights from our research team.
We would like to thank our patients and families for trusting us with their medical care during these challenging times. With the welcome news of emergency use authorization for COVID-19 vaccines, we look forward to 2021 with optimism and renewed confidence.

Pregnancy Outcomes in Late-Onset Pompe Disease
By Ozlem Goker-Alpan, MD
IN THIS OBSERVATIONAL CASE REVIEW STUDY, 25 FEMALE LOPD SUBJECTS, AGE 18 YEARS AND OVER WHO WERE PREGNANT OR WERE CONTEMPLATING PREGNANCY, WERE ENROLLED
Pompe disease (glycogen storage disease type II) is a rare lysosomal storage disorder resulting from the deficiency of the enzyme acid alpha-glucosidase. The phenotypic spectrum in Pompe Disease is varied, ranging from infantile, to juvenile and late-onset presentations.
Late-onset Pompe disease (LOPD) can present at any age, and primarily lower limb muscles, paraspinal muscles, and respiratory muscles are affected causing progressive weakness.
Enzyme replacement therapy (ERT) with alglucosidase alfa (Lumizyme®) was approved by the FDA in 2010 for the treatment of patients with LOPD. It has been initially classified by FDA as Pregnancy Category B but later changed to Pregnancy Category C, according to which potential benefits may warrant use of alglucosidase alfa in pregnant women despite potential risks.
In general, events acting as metabolic stressors, such as pregnancy are implicated as disease modifiers in metabolic disorders. However, the impact of pregnancy on LOPD is unclear. There is only limited information available as guidelines for clinicians in counseling women with LOPD who are planning to become pregnant.
LDRTC recently reported the results from a study specifically focusing on female patients with LOPD, to characterize the obstetric history and concomitant problems with Pompe Disease, exploring the disease progression in patients who have been pregnant, including the impact of ERT on the outcomes of pregnancy and the fetus (Goker-Alpan, et al., Life (Basel). 2020 Sep 11;10(9):194). In this observational case review study, 25 female LOPD subjects, age 18 years and over who were pregnant or were contemplating pregnancy, were enrolled at two centers. Findings for patients with LOPD were compared with national data from the Centers for Disease Control and Prevention (CDC) for the general population.
The impact of pregnancy on LOPD can be varied, while in the majority of patients it may not have an immediate clinical impact. Pregnancy may initiate symptoms of Pompe disease, leading to subsequent investigations and diagnosis, and in some patients, pregnancy may worsen symptoms. Only three patients reported respiratory compromise either with decreased exercise capacity or shortness of breath.
An important risk relevant to women with respiratory disease is one where maternal hypoxia results in oxygen saturations less than 85%, and the livebirth rate is only 12%. While complication rates in pregnancy or childbirth were not higher in study subjects, compared to the CDC’s National Vital Statistics Report, there was an increase in the rate of stillbirths (3.8%)
for the study subjects as compared to the national average (0.2–0.7%). However, these still births were found to be unrelated to Pompe Disease. Since only a minority of patients (3/25) reported significant pulmonary decline, we have ascertained disease progression through the use of assistive mobility devices. More than half of patients (14/25) were using an assistive device for mobility at the time of the study. Three patients reported worsening of symptoms in relationship to mobility, specifically during pregnancy.
In this study, two patients conceived while receiving alglucosidase alfa and one patient was started on ERT during pregnancy. There were no reports of adverse effects of ERT use in the four fetuses exposed during pregnancy. This cohort also demonstrates that patients and/or clinicians may have a preference to withhold therapy during a pregnancy. This decision could be based on the scarcity of data on the effects of ERT on the developing
fetus. One other concern could be the potential drug related immune hypersensitivity reactions. Infusion-related reactions, including anaphylaxis, urticaria, flushing, hyperhidrosis, chest discomfort, vomiting, and increased blood pressure were observed in up to 51% of the patients treated with alglucosidase alfa.
As discussed, LOPD is a progressive disorder, if untreated there is an expected decline both in peripheral muscle strength and respi- ratory function. Thus, the decision about ERT should be based on the individual patient, and special attention should be paid to already existing respiratory compromise, and prior history of immunogenicity against human recombinant alglucosidase alfa.
While there are certain limitations to the study, especially the small cohort size, the results are expected to help clinicians in counseling and caring for women with Pompe Disease, who are planning to become pregnant, and during the pregnancy.
Goker-Alpan et al., Life (Basel). 2020 Sep 11;10(9):194

Coping with Infusion Related Reactions in Fabry Disease Patients
By Renuka Limgala, PhD
Fabry disease (FD) is a rare genetic lysosomal storage disorder caused by mutations in the GLA gene and is inherited in an X-linked manner. It leads to a lack of or faulty ɑgalactosidase A (ɑ-Gal A) enzyme causing progressive damage that could lead to multi-organ failure involving kidneys, the heart, and the central nervous system.
Enzyme replacement therapy (ERT) with recombinant enzymes is the standard of care of treatment. It has proven to be successful in mitigating the pathological effects and improving the quality of life. However, infusion-related reactions (IRRs) are often seen in some FD patients as a result of the immunogenicity of infused exogenous enzymes.
The subsequent generation of antibodies in patients with no residual ɑ-Gal A activity can cause significant morbidity, leading to interruptions and occasional discontinuation of therapy. Interestingly, IgE antibodies usually associated with type 1 hypersensitivity reactions are often not found in FD patients with IRRs, suggesting that IgE-dependent immune pathways are not the only culprit for the most IRRs in FD.
The mechanisms and underlying immune perturbations resulting in hypersensitivity to the infused enzyme are not yet fully understood. In an attempt to better elucidate the role of the immune system in IRRs in FD patients, LDRTC has conducted a study on eight FD patients experiencing IRRs, comparing the results to FD patients who tolerate the ERT (Limgala et al., Am J Transl Res. 2019 Mar 15;11(3):1683-1696).
The immune profile showed no significant differences between FD patients with and without IRRs when pre- and post-infusion samples were compared; however, circulating dendritic cells were drastically reduced in reactors suggesting sequestration to the sites of inflammation. Recombinant proteins are thought to interact differently with dendritic cells from allergic and nonallergic patients, modifying their maturation level.
Dendritic cells can also metabolize the infused biological substances and present their metabolites to T cells resulting in a hypersensitivity response. It has been shown earlier that mast cell degranulation can activate dendritic cells to facilitate inflammatory responses. Toll-like receptor (TLR) pathways may play a role in mediating interactions between DCs, T lymphocytes, and mast cells, thus modulating allergic immune responses. Even though IgE antibodies were not found in the FD patients with IRRs, we wanted to investigate if mast cells are being activated in an IgE-independent manner leading to degranulation and resulting in the secretion of inflammatory cytokines.
Known mediators of mast cell degranulation—IL-4, IL-8, and TNF-ɑ—were quantified in plasma from pre- and post-infusion blood samples, which showed a significant increase of these cytokines, specifically in FD patients with IRRs, indicating nonspecific degranulation of mast cells in response to exogenous recombinant protein. Taken together, the results of this study indicate IgE-independent activation of mast cell degranulation, resulting in activating DCs and cross-talk with other immune players, including NK cells, as a possible mechanism for inflammatory responses causing IRRs in FD subjects .
In these patients, all IRRs were successfully managed using a combination of pre-medications and mast cell inhibitors (including mast cell stabilizers, steroids, and H1/H2 blockers) without any interruption of therapy. ADA titers were noted to decrease upon repeated exposure once ERT is tolerated. Mast cell stabilizers could thus be used to control nonspecific mast cell activation associated with IRRs. Further studies on such interactions might help in predicting the clinical outcome and developing strategies to target such interactions to counter IRRs.
Limgala, et al., Am J Transl Res. 2019 Mar 15;11(3):1683-1696
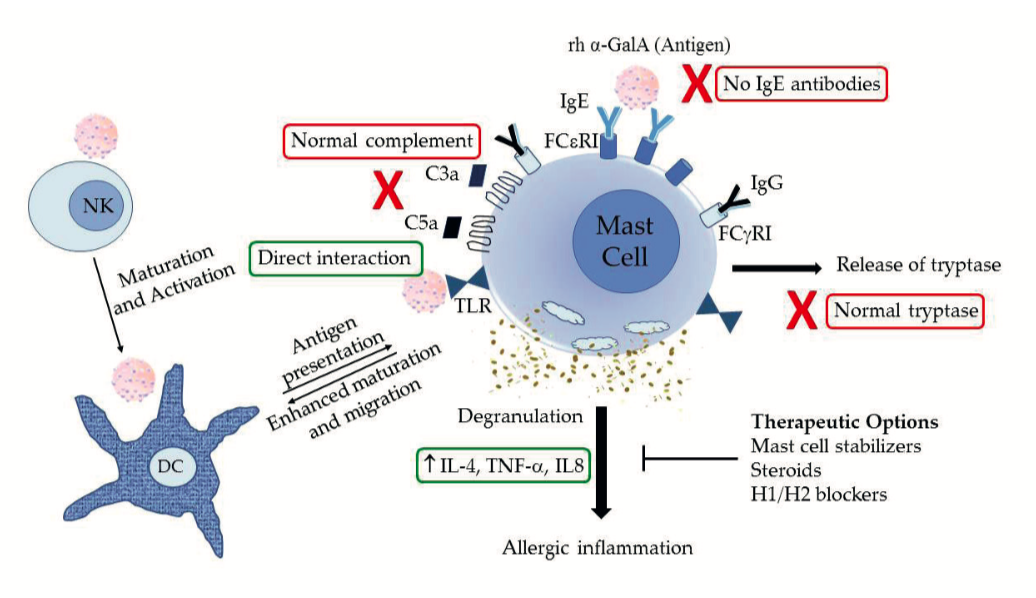
Possible mechanism of mast cell activation in irrs in Fabry disease patients. NK, natural killer cells; DC, dentritic cells; TLR, toll-like receptor; lg, immunoglobulin; rh ɑ-Gal A, recombinant human ɑ-galactosidase A (agalsidase); IL, interleukin; TNF, tumor necrosis factor.
Case Report: A Young Fabry Disease Patient with M296V GLA Variant and a Rapid Decline in Renal Function
By Ozlem Goker-Alpan, MD
Fabry disease (OMIM#301500) is an X-linked genetic disease caused by deficient activity of ɑ-galactosidase A (ɑ-Gal A). The lack of enzyme activity results in progressive accumulation of neutral glycolipids including globotriaosylceramide (Gb3) and globatriaosylsphingosine (Lyso-Gb3) in lysosomes of cells and body fluids. There are two clinical phenotypes, the classic type and the later-onset. Affected males with the classic form usually exhibit little or no ɑ-Gal A activity, and acroparesthesias, angiokeratomas, hypohidrosis, and corneal opacities in childhood or adolescence. With advancing age, the occurrence of renal disorders, cardiac disease, and stroke may lead to premature death in adulthood.
Here we present a case report for a young patient who was diagnosed with Fabry disease and inspite of treatment continued to experience worsening renal function. A 30-year-old patient was diagnosed with hypertension in 2016. His blood pressure readings at home were in the range of 130/90 mm Hg. He had a partial parathyroidectomy in 2010 due to recurring kidney stones.
A health screening in 2017 revealed “critical” BUN and creatinine levels and the patient was evaluated for nephrotic range of proteinuria of 1.4 gm of protein/day. The diagnosis of Fabry disease (Type 2 Late-onset) was made in 2017 by enzyme analysis and molecular testing, revealing the GLA variant M296V. His Fabry disease specific labs at that time were Lyso GL-3: BQL (ref. <0.30 ng/mL); Plasma GL-3: 2.5 μg/mL (ref. <7.0 μg/mL). M296 variant is located on the ß-strand (294–296) of the (ß/ɑ)8 barrel structure of GLA. It was reported that the amino acid substitution(s) at this location causes a small structural change localized to “the molecular surface”. Recalculation in this study revealed that the ASA value, RMSD value, numbers of atoms affected in the main chain, side chain and active site were 0 Å2, 0.018 Å, 5, 12, and 0, respectively. Affected atoms are located both in the core of GLA and on the molecular surface. These results suggest that M296 is buried and M296I causes a small structural change in small restricted region from the core to the surface, and it does not affect the active site. Thus, this may explain why the GLA variants at this site do not cause significant increase in the plasma Lyso-Gb3 level.
On examination there was no angiokeratomas, cornea verticillata, corneal clouding or clubbing. He had no hearing loss, cardiac involvement, no vascular or white matter abnormalities on his brain MRI/MRA. Patient began enzyme replacement therapy (ERT) with Fabrazyme at 1 mg/kg EOW soon after being diagnosed with Fabry disease.
In 2019, plasma GL-3 was 3.97 μg/mL (ref. 1.37-4.04 μg/mL). The patient’s kidney functions deteriorated rapidly over 24 months and he required dialysis. He developed stage 4 chronic kidney disease (CKD) and is currently being evaluated for renal transplant. He had a kidney biopsy and pathology showed severe arterial nephrosclerosis and predominantly sclerotic glomeruli. There was evidence of podocyte involvement with myelinosomes, which was concerning for Fabry disease. Further genetic evaluation with a Whole exome sequencing (WES) analysis was initiated to assess his rapid decli- ne in kidney function and atypical Fabry disease course with near normal biomarkers in an attempt to identify additional risk factor for rapidly progressive CKD. In addition to the known Fabry disease known pathogenic variants, WES identified a likely pathogenic VOUS (Variants of unknown significance) in NRLP3 gene, P882S.
Pathogenic variants of CIAS1/NRLP3 result in Muckle-Wells syndrome (MWS) is a dominantly inherited autoinflammatory disease characterized by fever, rash, arthralgia, conjunctivitis, sensorineural deafness and potentially life-threatening kidney amyloidosis. NRLP3 is suggested to mediate the inflammatory processes, and in renal biopsies of obtained from patients with nondiabetic kidney diseases, there was an increased expression of NRLP3 mRNA, which correlated with renal function, supporting the role for NRLP3 in renal injury. The NRLP3 genes are found to express in renal dendritic cells and macrophages, while other non-immune cells seem do not release IL-1ß. Novel inflammasome-targeting agents, including an IL-1ß monoclonal antibody, caspase 1 inhibitors and NRLP3 inhibitors, have shown promising effects in experimental models and there is current evidence that they may provide new therapeutic strategies for kidney disease by providing cardioprotection in patients with CKD.
In Fabry disease while the genotype is important as a prognostic factor, it is not the only determinant of the clinical course. A patient with Fabry disease who has an atypical presentation and/or course needs to be further evaluated for other genetic causes that may play a role.
Mitobe S. et al., Mol. Genet. Metab. 2012;107:623–626
Pan X. et al., PLOS One, 2016. https://doi.org/10.1371/journal.pone.0161330
Von Scheidt W. et al., New Eng. J. Med. 324: 395-399, 1991
Xiang H. et al., Front. Cell Dev. Biol., 2020. https://doi.org/10.3389/fcell.2020.00106
Current Safety Precautions Against COVID-19
The COVID-19 pandemic has forced all of us to adjust our habits when leaving the house. LDRTC has been implementing several safety measures to ensure our patients are safe and comfortable when visiting our clinic.
Here are the safety precautions taken by our facility:
• Monitor and follow CDC’s PPE recommendation
• Increased patient care with Telemedicine services
• Entire staff received vaccine against COVID-19
• Swab test for every patient that comes into the clinic
• Daily COVID-19 symptoms screening for staff and patients
• Private room for each patient and family
• Infusion rooms are fully disinfected after use
• Daily deep cleaning of the facility
• Hand sanitizers, disinfecting wipes, and gloves available
• Follow-up with patients who tested positive for COVID-19

Studies
Ambroxol Chaperone Activity in Gaucher Disease
By Margarita M. Ivanova, PhD
The study, “Individualized screening for chaperone activity in Gaucher disease using multiple patient- derived primary cell lines,” was published in the American Journal of Translational Research.
The effectiveness of ambroxol — a molecule that improves the folding and maturation of abnormal glucocerebrosidase — does not depend only on the type of GBA mutation in patients with Gaucher disease. Using blood cells from patients could help determine the treatment’s efficacy beforehand and improve personalized therapy approaches.
While enzyme replacement and substrate reduction therapies successfully treat the systemic manifestations of Gaucher disease, none of these approaches is effective at treating patients with the neuronopathic form of Gaucher disease. Gaucher treatments’ future might lie in small molecules — called chaperones — that bind the mutated glucocerebrosidase and help it fold properly. These molecules can cross the blood-brain barrier and treat Gaucher manifestations in the central nervous system; however, this molecule activity largely depends on the type of GBA mutation a patient has. Given that each patient has a unique genetic background, even patients with similar mutations might respond differently to these therapies. In other words, factors besides the GBA mutation itself would be involved in the response therapy, which would be difficult to predict based on the patient genotype only.
Ambroxol increased glucocerebrosidase activity in control cells inducing protein folding and its movement into the lysosome, where glucocerebrosidase exerts its function. However, not all cells with the L444P/L444P mutation leading to Gaucher disease type 3 — had an increase in glucocerebrosidase activity, suggesting that “ambroxol chaperone activity” could be modified by diverse factors and not only by the individual’s genetic variants.
In one project, we explored which cells could be used to determine whether a patient would benefit from ambroxol. Peripheral blood mononuclear cells— could lead to an expedited screening that took only five days.
Chaperone therapy is a promising approach for treating Gaucher disease, especially when the central nervous system is involved. Ambroxol demonstrated good tolerability while enhancing enzyme activity and improving neurological manifestations.
Ivanova MM, Erk Changsila, Alper Turgut, Ozlem Goker-Alpan. Am J Transl Res. 2018 Nov 15;10(11):3750-3761
Clathrin-Mediated Uptake of Recombinant ɑ-Gal A to Lysosome Activates Autophagy
The study, “Rapid Clathrin- Mediated Uptake of Recombinant ɑ-Gal A to Lysosome Activates Autophagy,” was published in the Biomolecules.
Enzyme replacement therapy (ERT) with recombinant alpha-galactosidase A (rh-ɑ-Gal A) is the standard treatment for Fabry disease.
ERT has shown a significant impact on patients; however, there is still morbidity and mortality in Fabry disease, resulting in progressive cardiac, renal, and cerebrovascular pathology.
The main pathway for delivering of recombinant enzyme to the lysosome is cation-independent mannose-6-phosphate receptor (CI-M6PR) endocytosis, also known as insulin-like growth factor 2 receptor (IGF2R) endocytosis.
One of our studies aims to investigate the mechanisms of uptake of rh-ɑ-Gal A in different cell types, with the exploration of clathrin-dependent and caveolin assisted receptor-mediated endocytosis and the dynamics of autophagy-lysosomal functions.
rh-ɑ-Gal-A uptake was investigated in primary fibroblasts, urine originated kidney epithelial cells, and peripheral blood mononuclear cells derived from Fabry patients and healthy controls. Uptake of rh-ɑ-Gal A was more efficient in the cells with the lowest endogenous enzyme activity.
Chloroquine and monensin significantly blocked the uptake of rh-ɑ-Gal A, indicating that the clathrin-mediated endocytosis is involved in recombinant enzyme delivery.
Alternative caveolae-mediated endocytosis coexists with clathrin-mediated endocytosis. However, clathrin-dependent endocytosis is a dominant mechanism for enzyme uptake in all cell lines.
These results show that the uptake of rh-ɑ-Gal A occurs rapidly and activates the autophagy-lysosomal pathway.
Ivanova M M, Dao J, KasaciN, Adewale B, Fikry J, Goker-Alpan O. Biomolecules. 2020 May 30;10(6):837.
Cardiac Energy Metabolism in Fabry Disease
By Margarita M. Ivanova, PhD
Cardiovascular pathology is a hallmark of Fabry disease. The deposition of Gb-3 and Lyso-Gb-3 inside the myocardium of Fabry patients affects the cardiac function, but secondary functional alterations also play a role in progressive cardiovascular pathology.
The importance of mitochondrial function for the maintenance of energy metabolism for normal cardiac function.
Machann et al. show that the impartment of mitochondrial function and energy metabolism plays a significant role in Fabry disease cardiomyopathy. Electron microscopic evaluation of cardiomyocytes from Fabry patient's hearts shows a decreased total area of mitochondria. Several clinical studies confirmed that dysfunction of cardiac energy metabolism and increased oxygen requirements via LV hypertrophy link to reducing ischaemic tolerance in Fabry patients.
How does a lysosomal enzyme deficiency translate into mitochondrial dysfunction in Fabry disease? Gb-3 accumulation induces ROS production by inhibiting of mitochondrial antioxidant SOD2 in vascular endothelial cells and human pluripotent stem cells derived from Fabry patients. Membrane lipid composition is altered in Fabry disease and directly affects the mitochondrial inner membrane. The mitochondrial respiratory chain complexes I, II, III, and IV are embedded in the mitochondrial membrane and could be affected by membrane lipid composition status.
Ivanova M M. JClinMed. 2020 Apr14;9(4):1116.
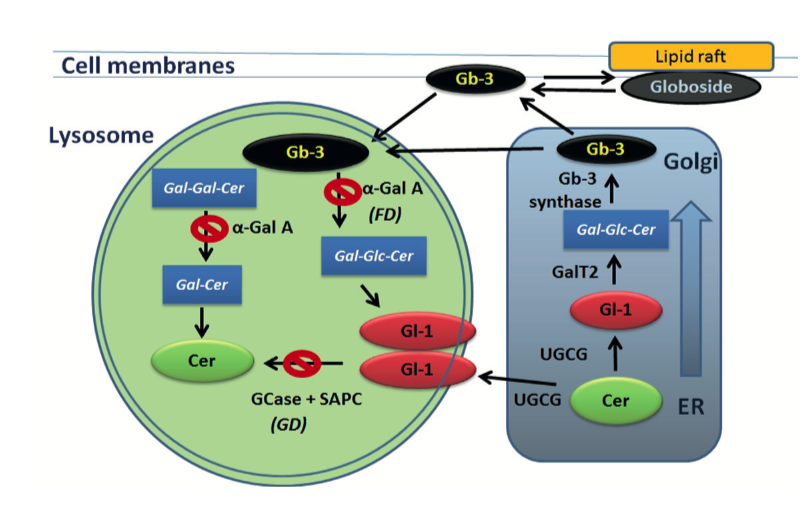
Sphingolipid metabolism in Gaucher and Fabry diseases. Ceramide (Cer), glucosylceramide (Gl-1) and globotriaosylceramide (Gb-3) are shifts between ER, Golgi apparatus, lysosomes and cellular membranes. UGCG synthase converts Cer to Gl-1 in the ER. Gl-1 localized in intralysosomal membrane is break down by GCase enzyme in present of SAPC. Gb-3 is synthesized from lactosylceramide (Gal-Glc-Cer) by Golgi-localized enzyme Gb-3 synthase; Gal-Glc-Cer is synthesized by LacCer synthase (GalT2) from Gl-1. Lysosomal accumulation of Gl-1 is link to GD. Lysosomal accumulation of Gb-3 is link to FD.
Role of Mitochondrial Function in Lysosomal Storage Disorders
By Margarita M. Ivanova, PhD
Sphingolipids represent a class of bioactive lipids that modulate the biophysical properties of biological membranes and play a critical role in cell signal transduction. Sphingolipids control crucial cellular functions such as the cell cycle, senescence, autophagy, apoptosis, cell migration, and inflammation. Sphingolipid metabolism is highly compartmentalized within the subcellular locations. However, the majority of steps of sphingolipids metabolism occur in lysosomes.
Altered sphingolipid metabolism with an accumulation of undigested substrates in lysosomes due to lysosomal enzyme deficiency is linked to lysosomal storage disorders.
Trapping of sphingolipids and their metabolites in the lysosomes inhibits lipid recycling, which has a direct effect on the lipid composition of cellular membranes, including the inner mitochondrial membrane. Additionally, lysosomes are not only the house of digestive enzymes but are also responsible for trafficking organelles, sensing nutrients, and repairing mitochondria. However, lysosomal abnormalities lead to alteration of autophagy and disturb the energy balance and mitochondrial function.
In this review, an overview of mitochondrial function in cells with altered sphingolipid metabolism was discussed, focusing on the two most common sphingolipid disorders, Gaucher and Fabry diseases.
Ivanonva M. J. Clin. Med. 2020, 9(4), 1116.
The review, “Altered Sphingolipids Metabolism Damaged Mitochondrial Functions: Lessons Learned From Gaucher and Fabry Diseases,” was published in the Journal of Clinical Medicine.
High Prevalence of Lysosomal Storage Disorders in Minorities of African Descent
By Renuka Limgala, PhD
LDRTC in collaboration with Howard University Hospital and College of Medicine, Washington DC, had initiated a large scale screening study for these LSDs to investigate the incidence of three LSDs in a cohort of mostly urban-dwelling African-Americans, compared to previous work primarily in Caucasians.
Lysosomal storage disorders (LSDs) are a group of about 50 inherited metabolic disorders usually resulting from mutations in the genes encoding lysosomal enzymes or enzymatic cofactors in the substrate degradation pathways.
The advent of enzyme replacement therapy followed by the development of substrate reduction and pharmacological chaperone therapies enabled different treatment alternatives for some of the common LSDs, including Gaucher (GD), Pompe (PD), and Fabry (FD). All three of these LSDs have juvenile or adult presentations with attenuated phenotypes. Varying disease onset and variable organ involvement complicate the diagnostic odyssey. As a result of treatment access, these disorders’ natural history has significantly changed, with a marked decrease in morbidity and mortality. However, it has been noted that delaying the initiation of therapy results in disease progression and complications, which then persist even after therapy has commenced. Hence, awareness and early diagnosis of these LSDs followed by prompt treatment play a vital role in alleviating the symptoms and stopping the disease’s progression.
Certain LSDs are known to affect people across various racial or ethnic groups. Indeed, population studies point to regional and ethnic differences, which must be considered in planning for healthcare service and delivery. The prevalence of these or any of the LSDs is yet to be determined in the minority/ethnic groups residing in the US.
LDRTC in collaboration with Howard University Hospital and College of Medicine, Washington DC, had initiated a large scale screening study for these LSDs to investigate the incidence of three LSDs in a cohort of mostly urban-dwelling African-Americans, compared to previous work primarily in Caucasians.
Study samples were obtained from a population with ~90% reported as African-American, ~5% Hispanic, and <5% Caucasian or other. For the GD-causing GBA gene, three subjects had either homozygous or heterozygous mutations. For the PD-causing GAA gene, 8 subjects were either homozygous or compound heterozygous for mutations. Three novel mutations: 1) c.472 A>G; p.T158A, 2) c.503G>T; p.R168L, 3) c.1985del were also identified in GAA. For the FD-causing GAA gene, two subjects had patho- genic mutations, and 4 had single nucleotide polymorphisms in the 5’UTR, previously implicated in modulating gene expression.
The findings highlight a higher incidence of abnormal enzyme levels and pathogenic mutations in the target population, reflecting ancestry-based specific genotype and phenotype variations.
The results from the large-scale undertaking in the current study provide much needed population data of GD, PD and FD prevalence in African-Americans. Despite some limitations of the study, given the fact that the diagnostic assays for these LSDs can be performed with relatively low cost and results can be confirmed using targeted single-gene sequencing, it would be advisable for the physicians responsible for the clinical care of these patients to be aware that these LSDs, which are considered rare diseases in the general population, may be more common in people of African ancestry and other minority groups. Underrepresentation of minority groups for both participation in clinical research trials and seeking specific medical care further hamper the clinical practice, specifically addressing the medical needs of patients of African descent.
Limgala R. et al., JIMD Reports (2021) doi.org/10.1002/jmd2.12201




Lysosomal Storage Disorders - Disease Mechanisms, and Patient Care and Management in the midst of COVID-19 crisis
LDRTC virtually hosted the 6th Genetic Rare and Immune Disorders Symposium (GRIDs) on the 1st of July 2020.
The summit gathered eighteen physicians, researchers, and patient advocates from Canada, Israel, Italy, the United Kingdom, and the United States. Speakers addressed the medical community and lysosomal storage disorder (LSD) patients’ challenges during the COVID-19 pandemic.
Experts in lysosomal and rare disorders presented data on disease mechanisms, patient care, and management amidst the health crisis.
Physicians from Europe and the US shared their insights about outpatient management, inpatient treatment, and long-term follow-up of SARS-CoV-2 infections in patients with these rare disorders.
During the event, participants had access to research addressing the effect of the virus on different treatment strategies for lysosomal diseases and how to manage enzyme replacement therapies with the COVID-19 high-risk patient population.
Physicians and researchers from LDRTC and other laboratories presented studies of SARS-CoV-2 infections and the pathogenic pathways in LSDs.
Researchers talked about the role and influence of cellular and lysosomal pathways and immunopatho-genesis in lysosomal disorders in COVID-19, susceptibility, severity, and the increased vulnerability
of multisystemic disorder patients COVID-19. GRIDs also highlighted the role of individual patients and patient advocacy groups in adapting to the pandemic induced changes in treatment strategies and the importance of maintaining communication with the medical community and industry professionals to receive timely updates.
Physicians, patient advocates, and researchers concluded the event by discussing the long-term impact of this pandemic on managing and treating rare diseases. “These shared experiences and lessons learned would serve as a way to establish much-needed guidelines to be better prepared for large-scale global events long after the COVID-19 pandemic has passed”, said Dr. Renuka Limgala, an LDRTC researcher.
LDRTC is at a New Location
3702 Pender Drive, Suite 170 Fairfax, VA 22030

The Lysosomal and Rare Disorders Research and Treatment Center (LDRTC) has moved to a new location in July 2020 to better accommodate our patients’ needs.
Our new facility combines patient care and two other branches - Clinical and Translational Research - in one place.
The new area offers a more spacious floor plan that includes six private infusion therapy rooms with reclining chairs, sofas, and TVs. The office also features two private exam rooms and onsite restrooms for our patients’ convenience.
Our clinic is equipped with an echocardiogram, EKG (electrocardiogram), and a PFT (Pulmonary Function Test) machine to better monitor patients when receiving care at our facility.
LDRTC also has an onsite Pharmacy, a Translational Research Laboratory, and a Conference room.
Our new address is 3702 Pender Drive, Suite 170, Fairfax, VA 22030
Come and visit us!
Our Services:
Infusions Injections
Physical Therapy
Evaluation
Genetic Screening
Consulting
You can reach us at:
Tel: + 1 703 261-6220
Fax: + 1 703 991-6592
